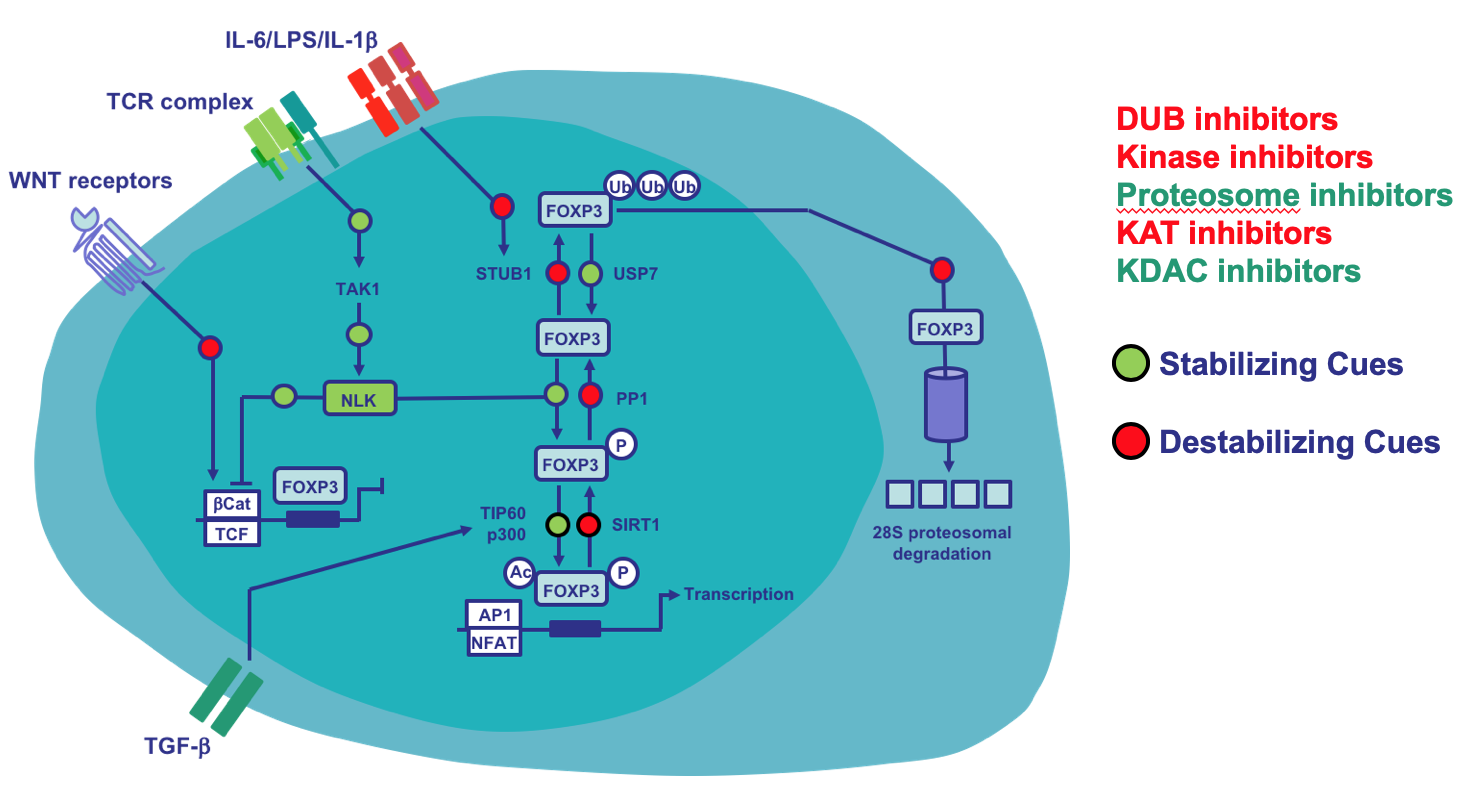
The Foxp3 transcription factor is a crucial determinant of both regulatory T (Treg) cell development and their functional maintenance. Appropriate modulation of tolerogenic immune responses therefore requires the tight regulation of Foxp3 transcriptional output, and this involves both transcriptional and post-translational regulation. Here, we show that during T cell activation, phosphorylation of Foxp3 in Treg cells can be regulated by a TGF-β activated kinase 1 (TAK1)-Nemo-like kinase (NLK) signaling pathway. NLK interacts and phosphorylates Foxp3 in Treg cells, resulting in the stabilization of protein levels by preventing association with the STUB1 E3-ubiquitin protein ligase. Conditional Treg cell NLK-knockout (NLKΔTreg) results in decreased Treg cell-mediated immunosuppression in vivo, and NLK-deficient Treg cell animals develop more severe experimental autoimmune encephalomyelitis. Our data suggest a molecular mechanism, in which stimulation of TCR-mediated signaling can induce a TAK1-NLK pathway to sustain Foxp3 transcriptional activity through the stabilisation of protein levels, thereby maintaining TREG cell suppressive function. This work is a collaborative effort between our group and Dietmar Zaiss and has been driven by Veerle Fleskens (UMCU) and Carlos Minutti (University of Edinburgh) together with many important international partners. Part of a long-term research line we have identified a druggable Foxp3 stability circuit that could be used to modulate Treg cell activity in a variety of pathological situations. You can read more about this work here in Cell Reports.